La IHH (hipogonadismo hipogonadotropico idiopatico) puede ocurrir con olfacción normal (IHH normósmica) o con anosmia. Esta última presentación clínica de IHH con anosmia se conoce como síndrome de Kallmann (KS).
Los pacientes con IHH a menudo tienen características clínicas que están presentes al nacer (criptorquidia, micrófalo y/o evidencia bioquímica de bajas gonadotropinas y esteroides sexuales durante los tres a seis meses del período neonatal denominado «mini-pubertad»).
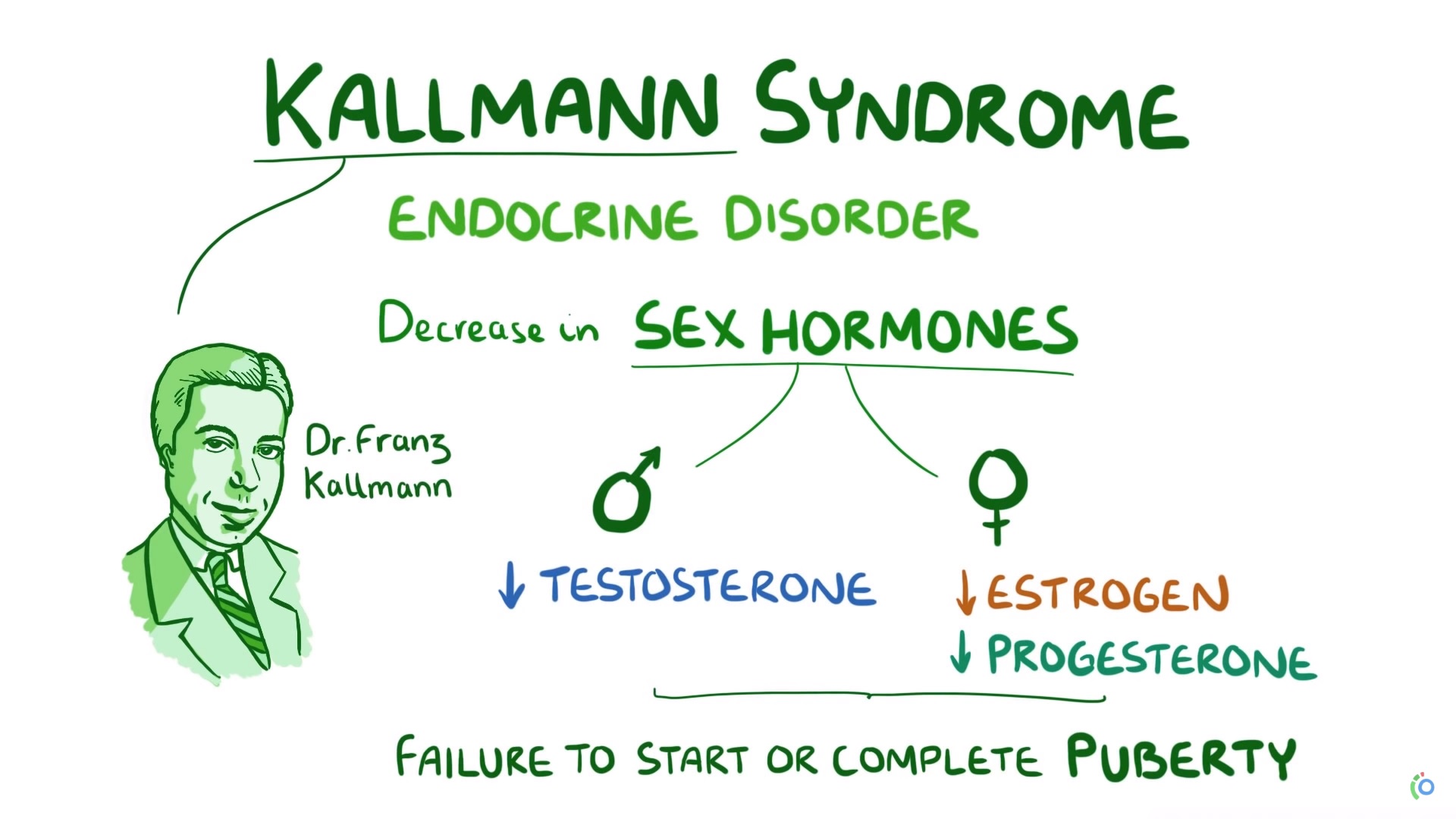
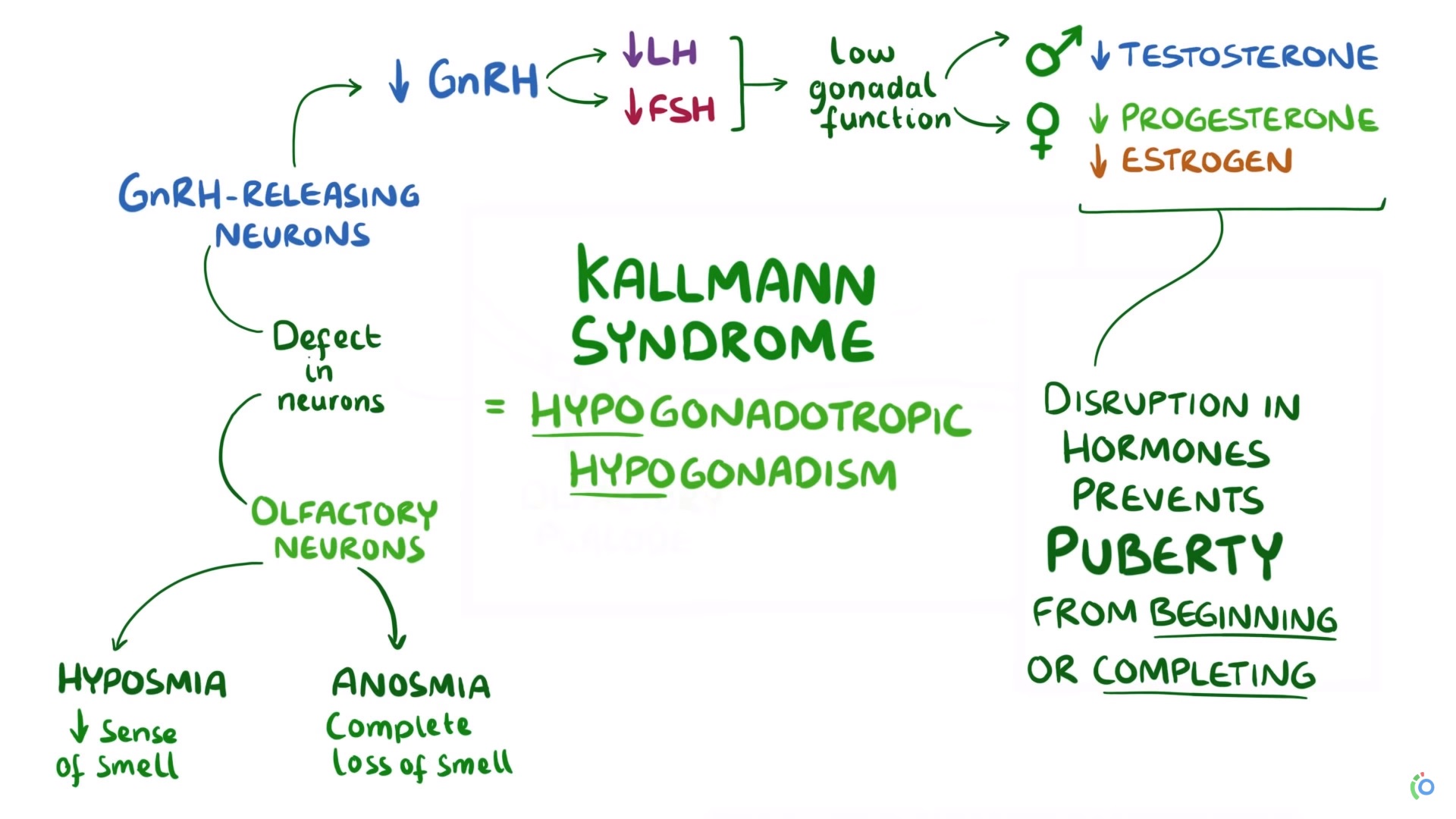
Forma anósmica de IHH (síndrome de Kallmann [KS])
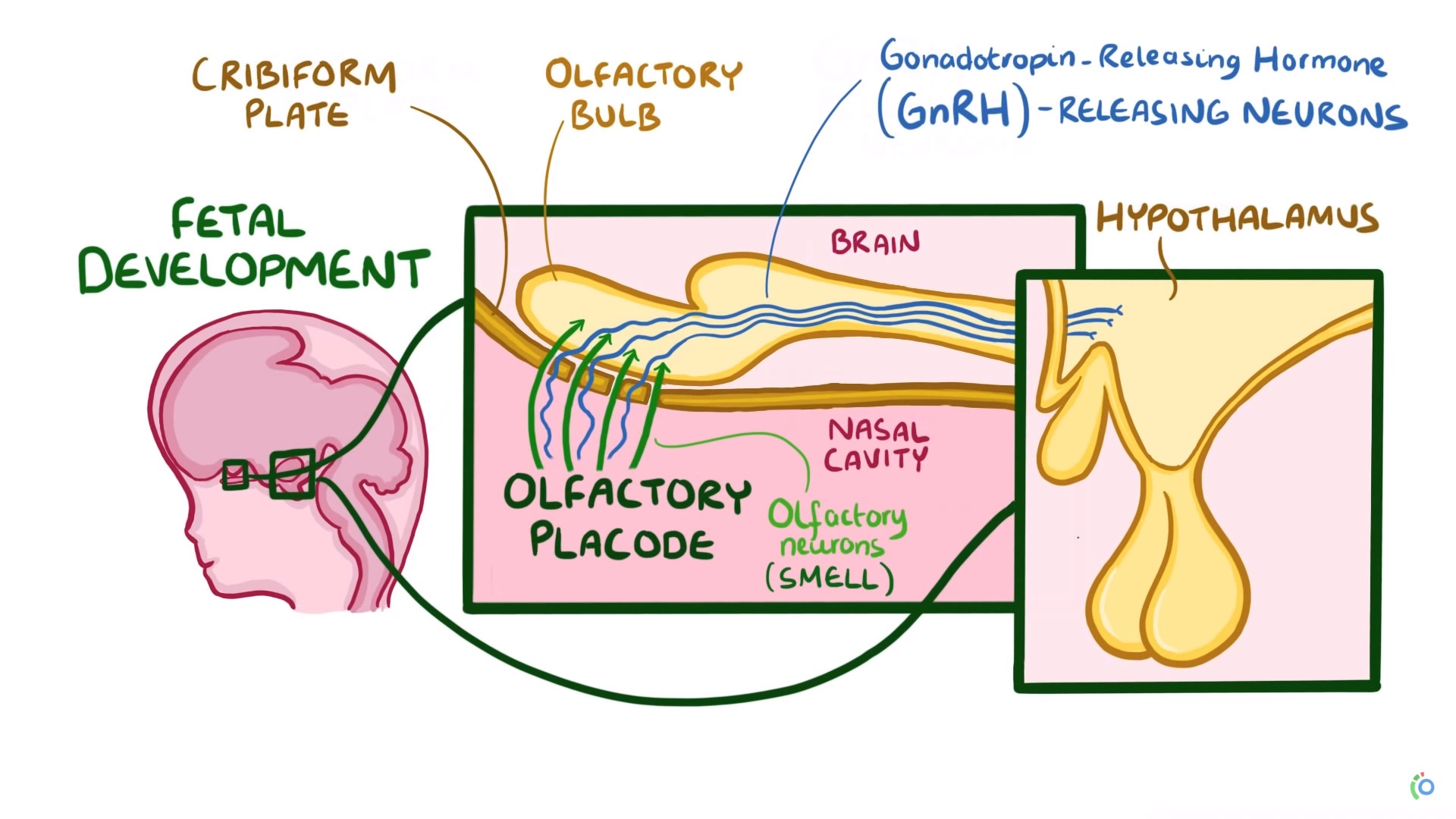
ANOS1 (anteriormente KAL1 ) ubicado en la región Xp22.3 del cromosoma X, fue el primer gen encontrado que se mutó en KS. A diferencia de otras neuronas neurosecretoras hipotalámicas, las neuronas GnRH se originan en el epitelio olfativo, que se encuentra fuera del cerebro en desarrollo, y luego migran hacia el hipotálamo en desarrollo durante la vida fetal. La migración adecuada de estas neuronas depende de la expresión correcta de anosmin-1, una proteína que está codificada por el gen ANOS1.
Cuadro Clínico
La expresión clínica que puede ocurrir en el IHH, que va desde la ausencia completa de desarrollo sexual hasta la finalización parcial de la pubertad que no progresa posteriormente.
El IHH puede presentarse a cualquier edad, pero los signos y síntomas que se presentan son una función del período de actividad reproductiva relacionado con la edad.
- Durante el período neonatal, los niños con los casos más graves de IHH pueden presentar micrófalo y / o criptorquidia, probablemente debido a deficiencia de GnRH en el útero y / o neonatal. Las niñas recién nacidas con IHH no tienen hallazgos anormales obvios del tracto reproductivo que puedan proporcionar pistas para el diagnóstico. Sin embargo, en ambos sexos, otras características congénitas no reproductivas pueden estar presentes (por ejemplo, defectos faciales en la línea media, anomalías esqueléticas).
- En la pubertad, los pacientes de ambos sexos pueden presentar una forma completa de IHH que se caracteriza por no iniciar la maduración sexual (p. Ej., Falta de características sexuales secundarias, amenorrea primaria en las niñas, falta de virilización en los niños).
En las mujeres, las características sexuales secundarias a menudo están completamente ausentes, con poco o ningún desarrollo mamario o vello axilar.

Varias anomalías congénitas se pueden encontrar en los pacientes con IHH, en particular los que tienen la forma de KS IHH, debido a su etiología del desarrollo.
- Defectos en la línea media (es decir, labio / paladar hendido)
- Anosmia / hiposmia
- Agenesia renal unilateral
- Criptorquidia uni o bilateral
- Sincinesia bimanual (o movimientos espejo)
- Sindactilia u otras anomalías esqueléticas
- Pérdida de audición
- Agenesia dental

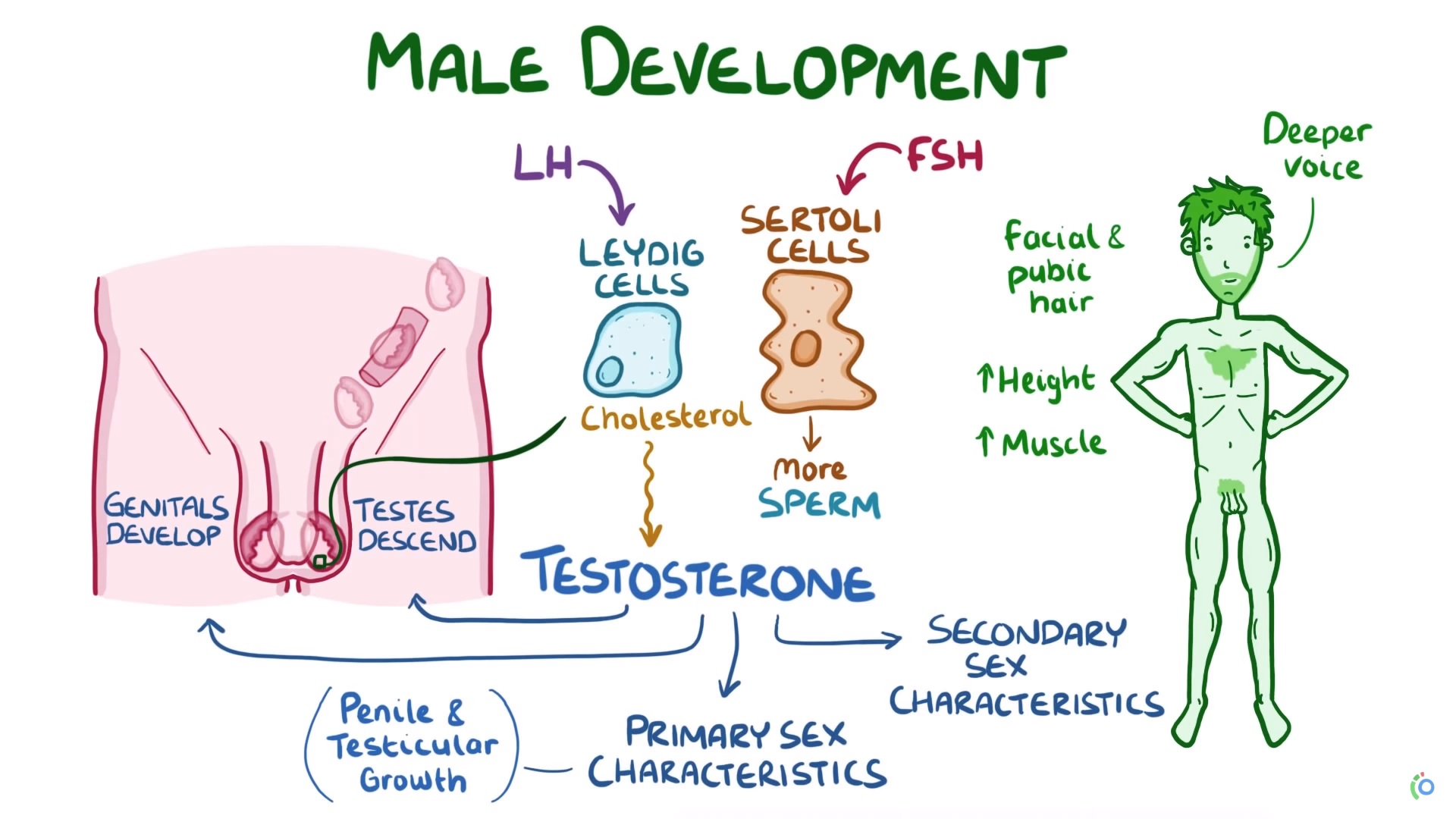
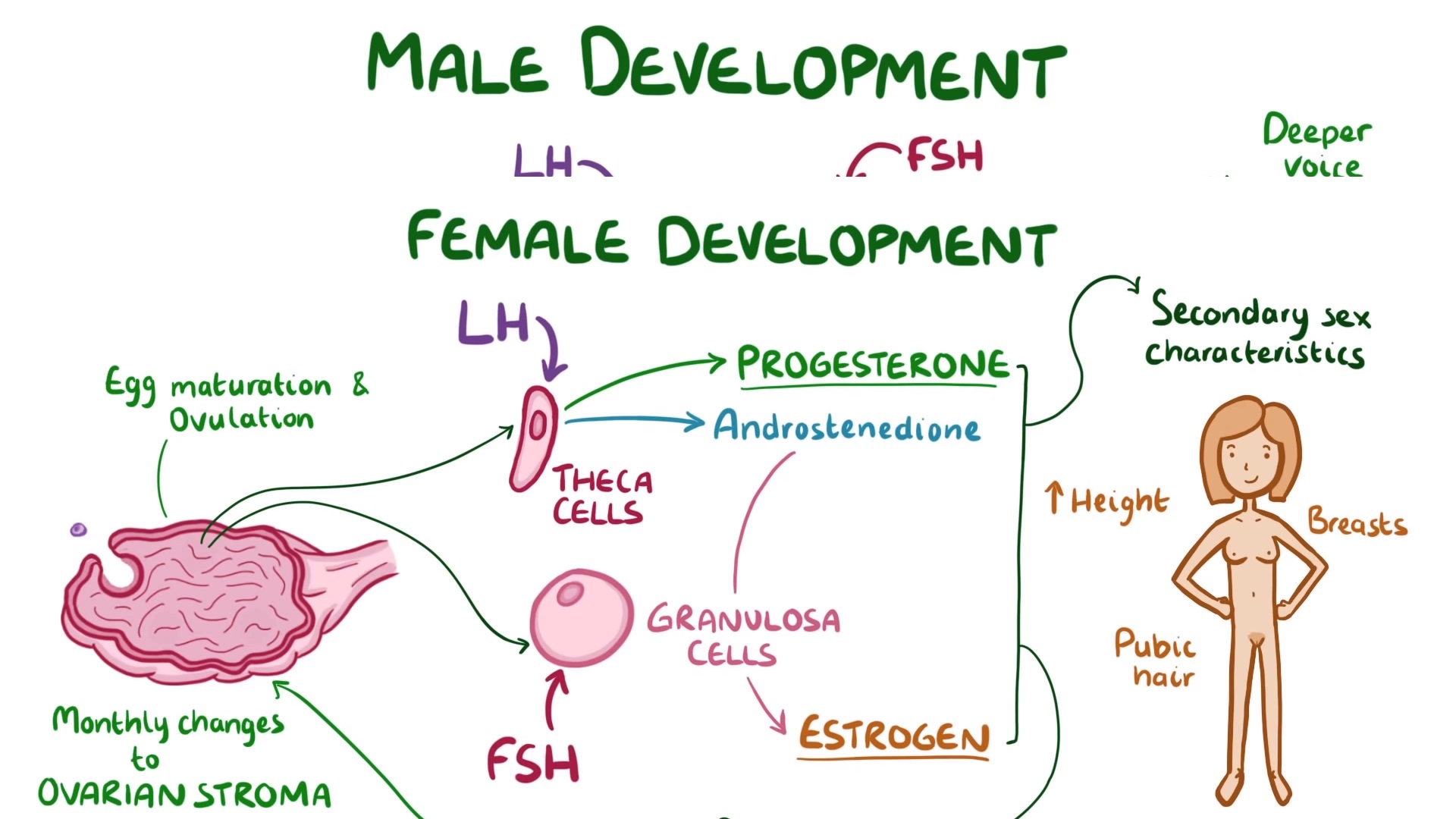
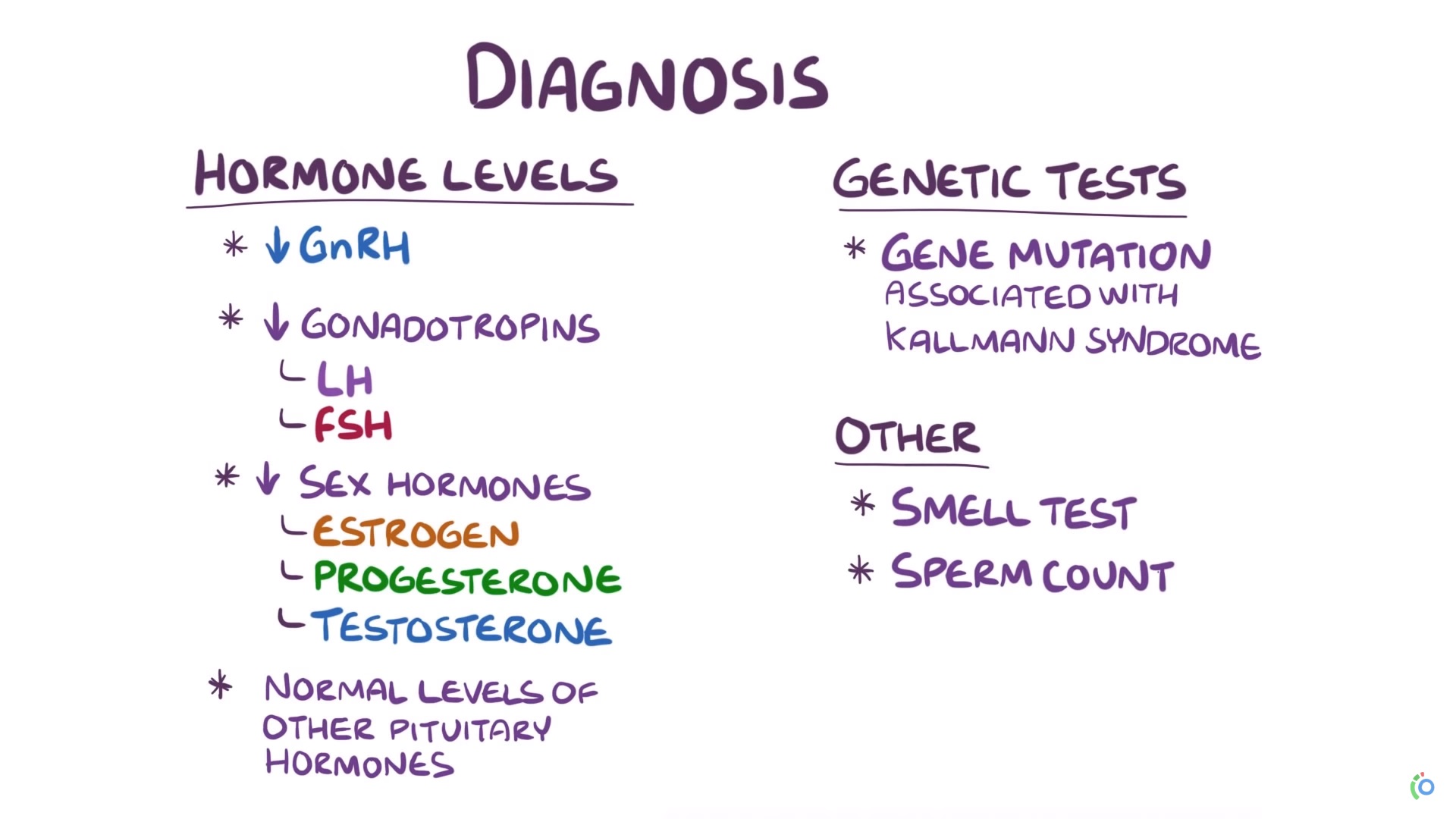
Diagnóstico
- La demostración de concentraciones séricas prepuberales de hormonas esteroides sexuales (testosterona sérica <100 ng / dL en hombres o estradiolsérico <20 pg / ml en mujeres).
- Concentraciones de LH y FSH bajas o normales en lugar de las concentraciones altas esperadas con la falla gonadal primaria
- Hipotálamo e hipófisis normales a la IRM
Para los pacientes que cumplen con los criterios de laboratorio anteriores, el diagnóstico diferencial principal (y el más difícil) es el retraso constitucional del crecimiento.
NOTA: Es difícil realizar un diagnóstico definitivo de IHH en ausencia de antecedentes familiares o pruebas genéticas previas hasta que el paciente cumpla al menos 18 años de edad, a menos que haya otras características sugestivas (es decir, micrófalo y / o criptorquidismo previo , anosmia, agenesia renal, defectos esqueléticos, etc).
Ninguna prueba individual puede distinguir de manera confiable entre IHH y retraso constitucional del crecimiento hasta que se disponga de pruebas genéticas más amplias, y por lo tanto, uno tiene que confiar en una serie de pistas clínicas, así como en la evolución natural en el tiempo. Sin embargo, ciertas características pueden indicar una mayor probabilidad de IHH en lugar de CDGP:
- Un historial familiar de deficiencia de la hormona liberadora de gonadotropina (GnRH), anosmia y / o la presencia de una o varias anomalías congénitas asociadas sugiere anomalías congénitas no reproductivas (Labio leporino / paladar, sindactilia) sugiere una forma de KS de deficiencia de GnRH.